不可能的任務?
針對這個國人關心的問題,陳垣崇表示,15年前或許不可能,但如今台灣的生醫水準已經具備研發新藥的能力,關鍵是,從研發成功到量產上市,仍有漫長而艱困的路要走,若不能堅持到底也是枉然。
主要的障礙,在於現行醫藥法規對於人體實驗的種種限制。人體實驗是新藥上市前必經的階段,在通過動物實驗、進行人體實驗前,必須向財團法人「醫藥品查驗中心」提出申請,然而,我國一直以來有個不成文的規定──通過美國藥物食品管理局(FDA)核可者,就能獲得快速審核,拿到衛生署藥政處核發人體實驗的許可。若未事先拿到此一「背書」,醫藥品查驗中心將會對新藥人體實驗的風險性、有效性、道德性等條件逐一審核,曠日廢時。
5月初,中研院院長李遠哲在Myozyme發表記者會上表示,過去台灣研發新藥實力不足時,需處處仰賴外國的查驗鑑定,如今學界、業界的能力已經提升,政府管理的配套措施也應該有所調整。
此外,現行的醫療法規對於醫療糾紛,除了民事賠償外,醫師還必須負擔刑事責任。對於結果難以預料的新藥人體實驗來說,無疑是相當大的風險,萬一實驗失敗,病人可以一狀告上法庭,醫生除了賠錢外還得坐牢。
在這樣不利的情勢下,台灣醫生難免對配合研發團隊進行臨床人體實驗有所顧忌,也大大阻礙了生藥科技的發展。
終極目標:基因治療
新藥研發成功,過去束手無策的「龐貝氏症」有了解藥,台大醫院隨即首開全球先例,為新生兒進行龐貝氏症篩檢。
陳垣崇指出,台大醫師胡務亮最近就篩檢出2名龐貝氏症兒,有幸能在出生第20天就開始給藥治療,將傷害減到最低。
陳垣崇研發龐貝氏症解藥的階段性工作已經完成,但卻未達最終目標。他指出,病童雖然有藥可治,但必須終其一生接受治療,即使目前健保已納入給付,對病患來說,時間和金錢仍是沈重的負擔,未來唯有發展基因治療,才能一勞永逸。
和其他疾病的基因治療研究相同,龐貝氏症也是卡在基因殖入患者體內後「存活不久」和「表現不佳」的兩大難題上。
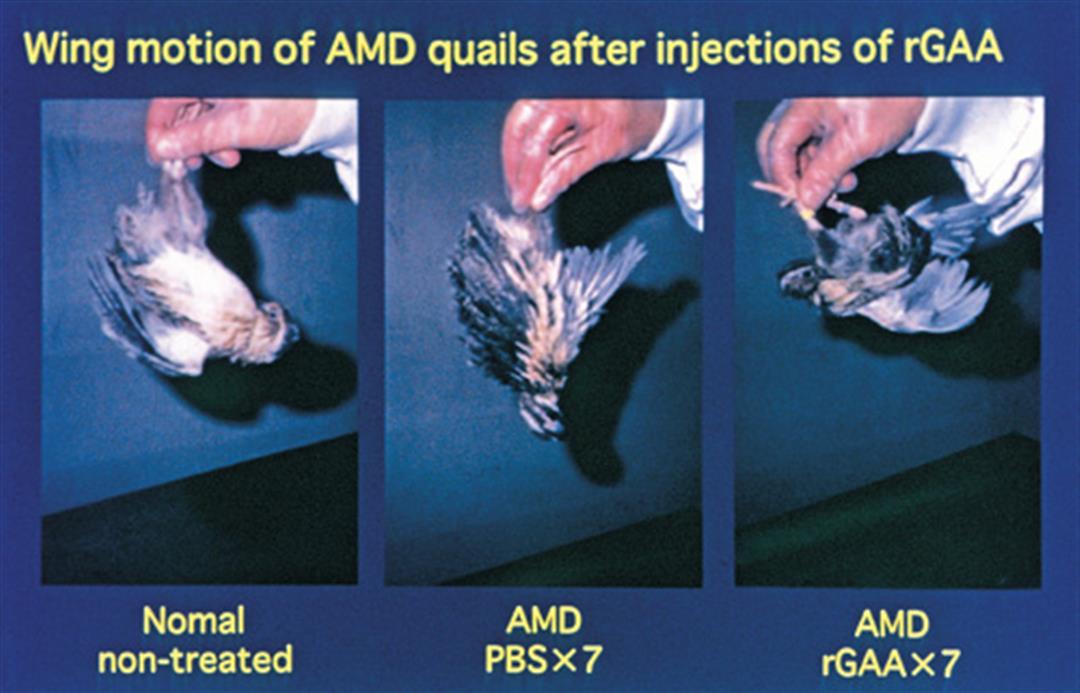
「目前動物實驗已經可以成功地置入健康基因以取代缺損的基因,問題是外來基因置入後會遭到患者自體免疫細胞的攻擊,大約維持6個月左右就弱化而消失不見了,」陳垣崇坦承,龐貝氏症的基因治療還有一段路要走。
除了龐貝氏症外,在陳垣崇帶領下,中研院生醫所還分別與國內各大醫學中心合作,研究包括糖尿病、躁鬱症、年輕型高血壓、手部退化性關節炎、肥胖等非單一基因引起、外加環境因素的疾病,以及12種藥物不良反應計畫。這些罹病人口眾多、市場龐大、影響深遠的疾病,是未來幾年生醫所致力的方向。
Myozyme只是一個開端,但它已為台灣生醫科技譜出序曲,後續的篇章值得期待。
龐貝氏症小檔案:
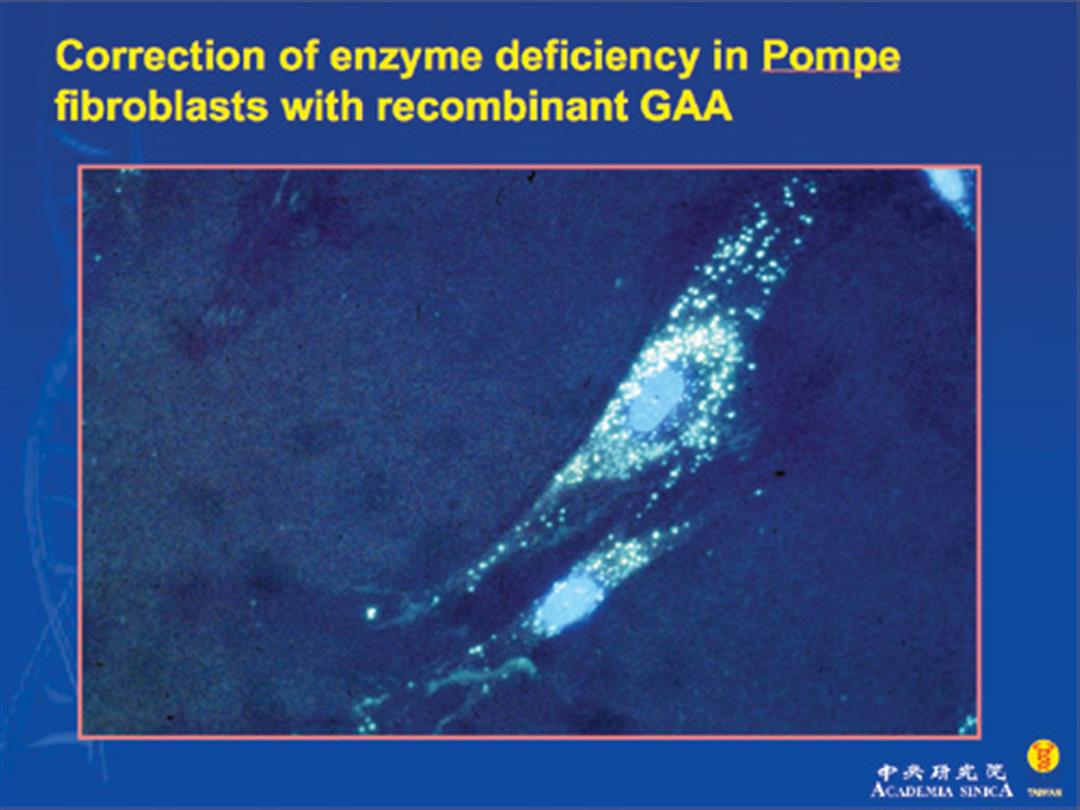
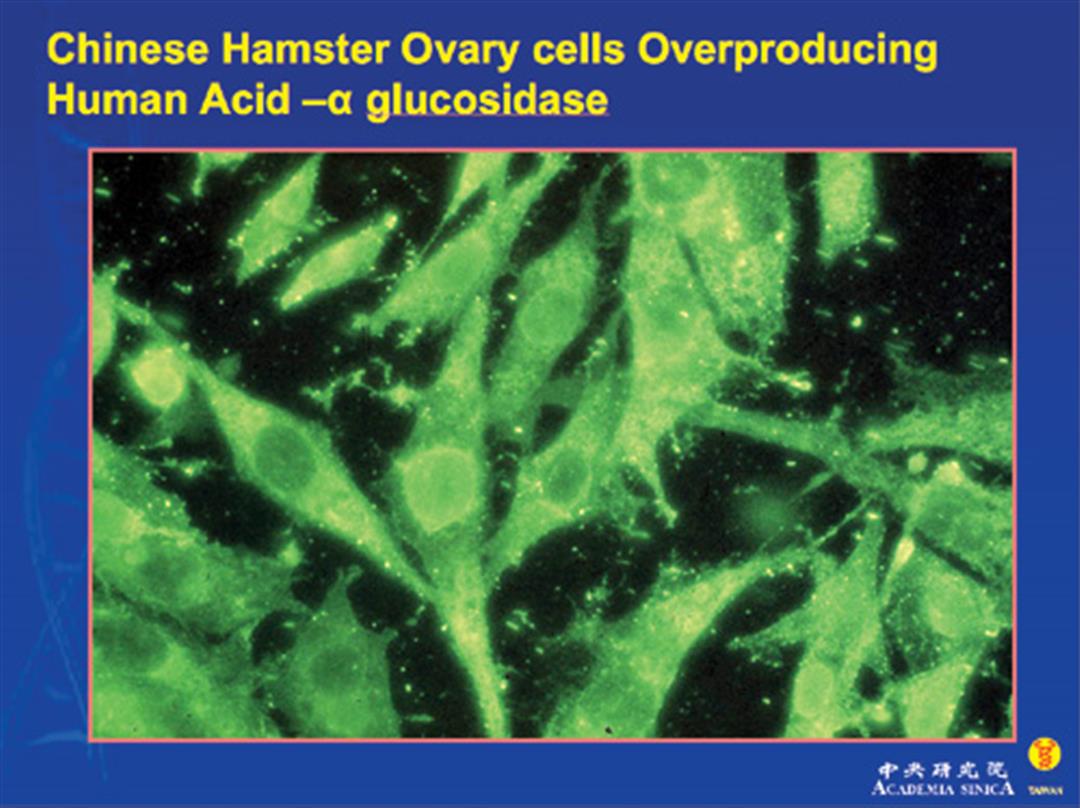
龐貝氏症是一種罕見且嚴重的遺傳疾病,又稱為「酸性麥芽糖酵素缺乏症」或「肝醣儲積症」。患者因先天缺乏分解肝醣的酵素,使得多餘的肝醣堆積在體內,傷害全身肌肉功能。
龐貝氏症確實的盛行率未知,種族間亦有所差異,粗估約4萬名新生兒中有一人罹病。
龐貝氏症因發病年齡不同,分為「嬰兒型」及「晚發型」兩種,其中嬰兒型較為嚴重,常危及生命,罹病病童因肌肉無力無法行走,被稱為「趴趴兒」。
「晚發型」則從兒童時期到晚年都可能發生,其惡化較慢,個別差異很大,有人只是肌肉無力,有人則需要輪椅或呼吸器。
龐貝氏症兒的症狀有:嚴重肌肉無力、心臟肥大、舌頭肥大、肝臟肥大、呼吸困難,常在發病第一年死於心臟呼吸衰竭。晚發型的症狀主要在肌肉無力,行走及呼吸困難,心臟的功能通常是正常的。
生醫所未來研究重點
中研院生醫所主要的研究對象是人類的疾病,先後建立了8個小組:
.癌症研究組
.心臟血管研究組
.感染疾病研究組
.神經科學研究組
.流行病學與遺傳研究組
.結構生物學研究組
.細胞與訊息傳遞研究組
.生物資訊研究組
生醫所累積多年的研究成果,將疾病與基因體研究接軌是未來的努力方向。
未來5年內,生醫所的「基因體醫學研究」重點分以下3個階段:
致病新穎基因與標的的篩選
致病基因功能的鑑定與分析
探討基因作為治療疾病的工具









@List.jpg?w=522&h=410&mode=crop&format=webp&quality=80)